What Causes Craniosynostosis?
While there is a known genetic component to craniosynostosis, the majority of cases are sporadic (meaning that they arise without an obvious genetic predisposition). Craniosynostosis can run in some families, and is seen in conjunction with certain craniofacial anomalies such as: Crouzon’s, Apert’s, and Kleeblattschadel syndromes.
How is Craniosynostosis Diagnosed?
In many instances a child’s parents or pediatrician simply notice an abnormal shape to the child’s skull. Other times it is diagnosed as part of the more complex craniofacial syndromes mentioned earlier. Rarely craniosynostosis is first diagnosed when a child presents with developmental delay.
The key to diagnosing craniosynostosis is differentiating it from a phenomenon called positional molding, which can have a similar appearance. Positional molding results when an infant develops a preference for resting on one side of the head, leading to an asymmetric flattening of that side of the skull. Frequently craniosynostosis can be diagnosed on physical exam alone. Occasionally x-rays of the skull and/or CT scans of the brain and skull are helpful in confirming the diagnosis.
How is Craniosynostosis Treated?
While cases of “positional molding” can be treated with helmets and/or adjusting the patient’s positions while resting, craniosynostosis is treated surgically. Surgery usually involves removing a portion of the skull, permitting the boney plates that make-up the rest of the skull to shift and assume a more normal contour.
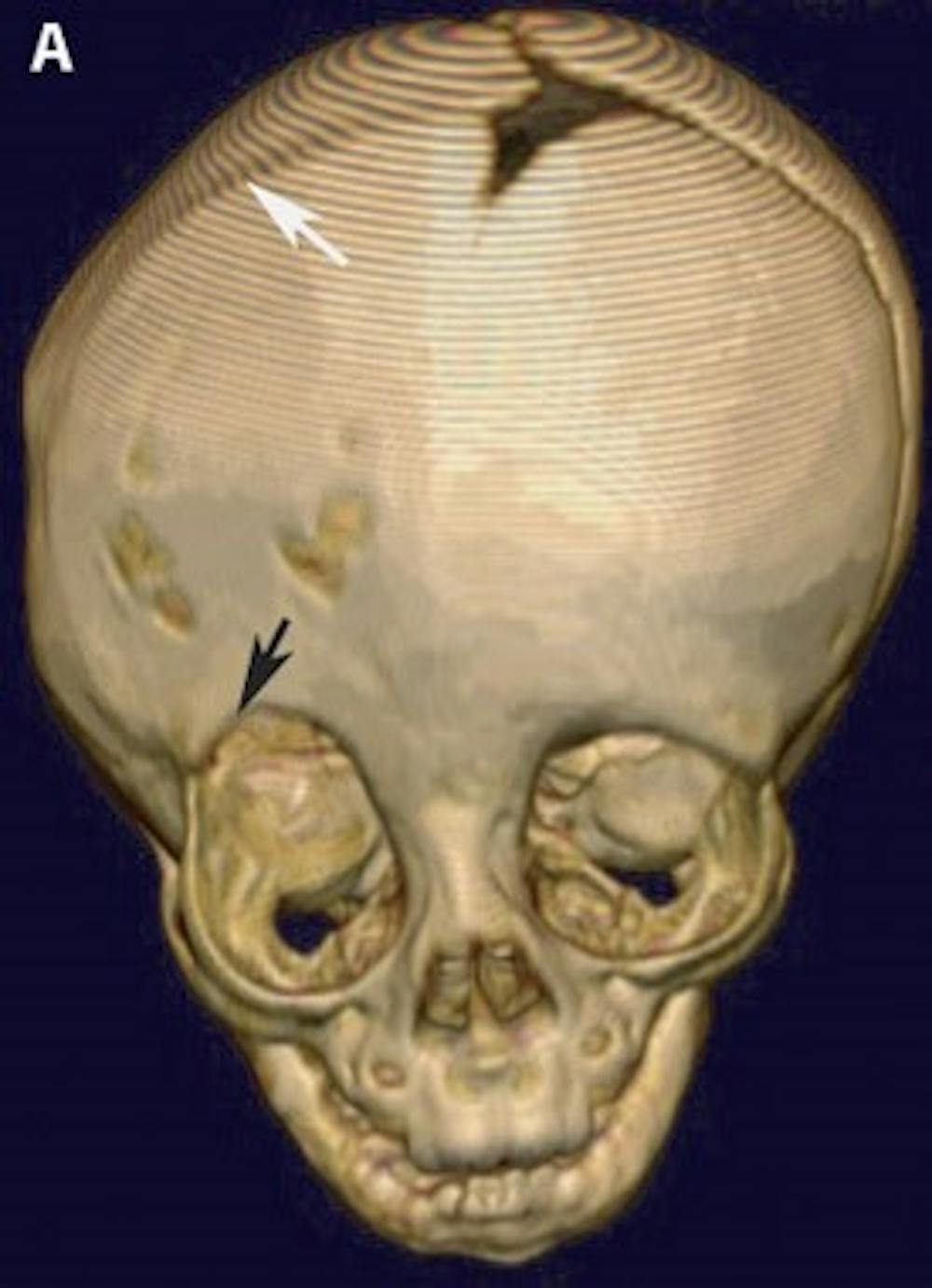
A) Pre-operative 3-D CT reconstruction of the skull demonstrating the effects of craniosynostosis. Note how the premature fusion of the right coronal suture (white arrow) has led to a distortion of the overall shape of the skull and right orbit (black arrow)
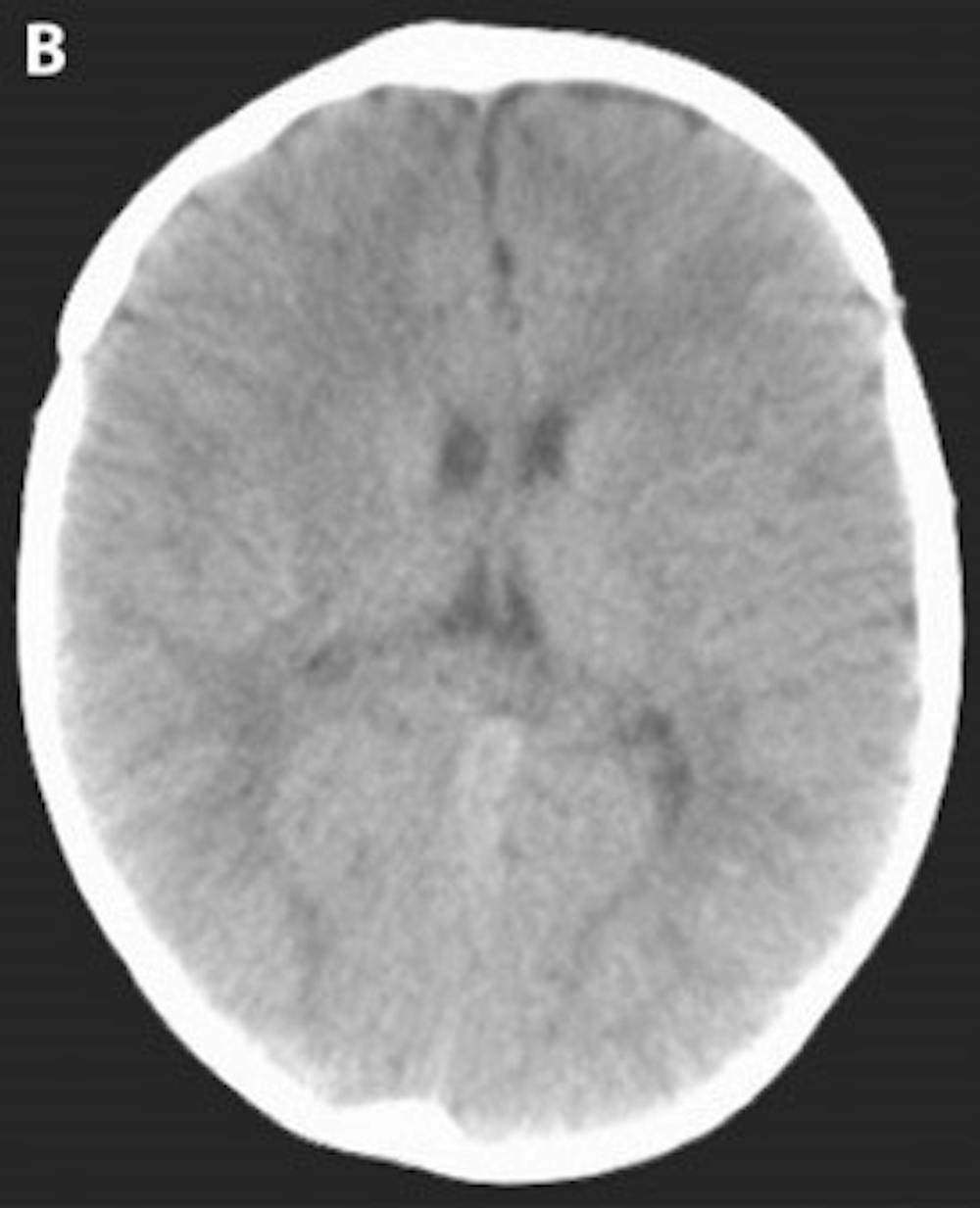
B) Post-operative head CT demonstrating a more cosmetically normal, rounded appearance to the skull